两电子氧化在合成和催化中无处不在,并起着举足轻重的作用。对于过渡金属和锕系元素来说,两电子氧化常发生在单个金属中心,如氧化加成反应和氧原子转移反应等(图1a)。然而,由于缺乏可变氧化态,稀土元素的氧化反应局限于单电子过程,如常用的单电子还原剂SmI2和单电子氧化剂(NH4)2[Ce(NO3)6],而两电子过程则需要两个或以上金属中心的参与(图1b)。近年来,稀土元素非寻常氧化态化学的发展为实现单一稀土元素中心的两电子过程提供了可能。但是,非正三价稀土离子的不稳定性以及不同价态稀土离子间的巨大差异阻碍了这一目标的实现。近日,北京大学化学与分子工程学院黄闻亮课题组利用三脚架型三胺基芳烃配体成功稳定了铈的三种氧化态(+2到+4价),并首次实现了从Ce(II)到Ce(IV)的两电子氧化(图1c)。该成果以“Two-electron oxidations at a single cerium center”为题,于2023年9月22日在《美国化学会志》(Journal of the American Chemical Society)上在线发表,并被选为封面文章。
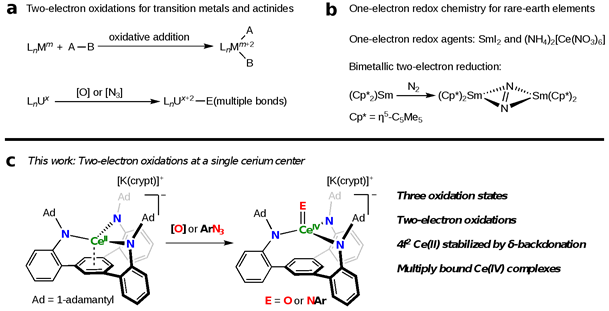
图1:稀土元素与过渡金属和锕系元素氧化还原化学的比较
该课题组近年来基于“f区金属–芳烃协同作用”的概念,发展了一类三脚架型三胺基芳烃配体并将其引入f区金属配位化学(图2a)。最近,利用金刚烷基取代的配体((AdTPBN3)3‒)来稳定铀的五种氧化态(+2到+6价),并证实了底座芳环能够起到同时稳定低价和高价铀离子的作用(图2b)。在此工作中,作者利用该配体稳定铈的三种氧化态,+2到+4价,并首次实现了从Ce(II)到Ce(IV)的两电子氧化反应(图2c)。
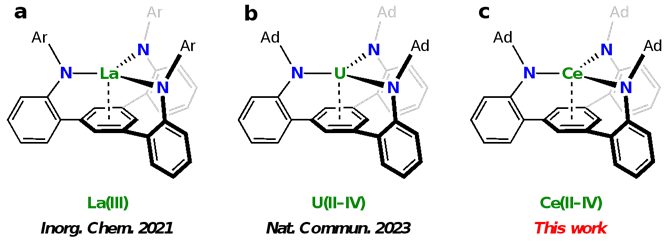
图2:三脚架型三胺基芳烃配体支撑的f区金属配位化学
从配体钾盐K3(AdTPBN3)和三价铈前驱体CeX3(THF)4 (X = Br or I)出发,通过盐复分解反应可得三价铈的配合物(AdTPBN3)Ce(III) (1)。电化学测试结果显示,1在‒2.68 V (vs. Fc+/Fc)处有一个可逆的还原峰,而在‒0.26 V (vs. Fc+/Fc)则有一个不可逆的氧化峰,表明1的化学还原和氧化是可行的。用KC8/crypt(穴醚222)还原1可以得到二价铈的配合物[K(crypt)][(AdTPBN3)Ce(II)] (K[2])(图3a)。核磁氢谱显示,2中底座芳烃的氢位于极高场‒178 ppm,表明铈与底座芳烃存在较强的轨道相互作用。尽管1与一些常用氧化剂无法得到明确的结果,但利用TeCl4氧化后再加Me3SnF处理的方法,可以得到稳定的四价铈的氟化物(AdTPBN3)Ce(IV)F (4);此外,1与KOtBu反应后再通入氧气可以得到四价铈的叔丁氧配合物(AdTPBN3)Ce(IV)OtBu (5)(图3b)。这些结果表明(AdTPBN3)能够稳定铈的三种氧化态(+2到+4价),为进一步尝试从二价铈到四价铈的直接两电子氧化打下了基础。K[2]与吡啶氧化物或3,5-双(三氟甲基)苯基叠氮(N3ArCF3)反应,能够得到四价铈的末端氧配合物(AdTPBN3)Ce(IV)O (K[7])或末端亚胺配合物(AdTPBN3)Ce(IV)NArCF3 (K[8])(图3c)。这是首个稀土元素的单中心两电子氧化反应。
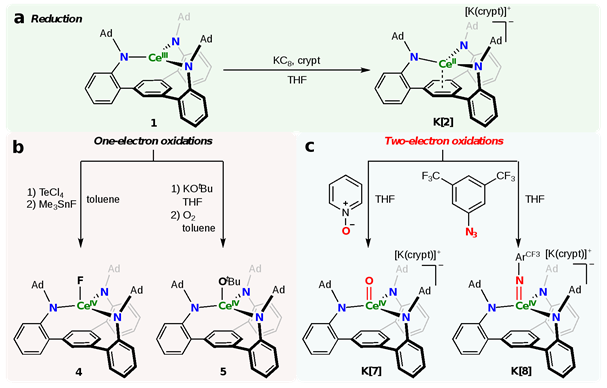
图3:铈配合物的合成及其氧化还原化学:a 三价铈还原为二价铈;b 三价铈的单电子氧化反应;c 二价铈的两电子氧化反应
作者对铈配合物进行了全面的谱学、结构和磁性表征,包括核磁共振波谱、紫外‒可见‒近红外吸收光谱、红外光谱、单晶X射线衍射、超导量子干涉仪磁学(SQUID)和电子顺磁共振(EPR)的表征,来探究其电子结构。二价铈配合物K[2]的变温磁化率测试表明,在低温下其磁矩趋近于零,符合4f2电子构型对应的3H4非磁基态;但在室温下其磁矩显著低于“自由4f2”的三价镨离子,表明4f轨道可能参与成键。低温EPR测试显示,K[2]的固体样品没有EPR信号,而冻溶液样品仅有微弱的可归属于溶剂化电子的信号,排除了K[2]为三价铈离子和芳烃自由基阴离子的可能,进一步支持K[2]为4f2电子构型的二价铈配合物。
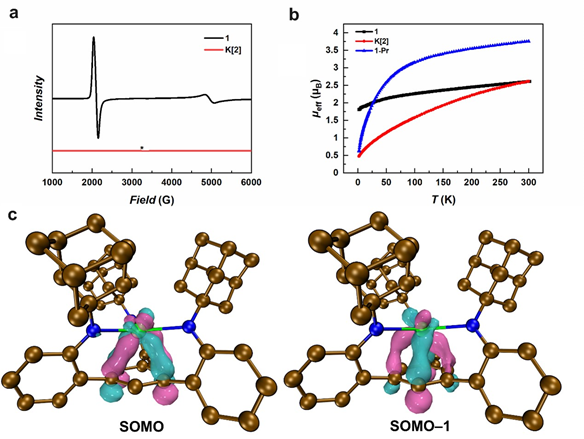
图4:二价铈配合物K[2]的电子结构:a1和K[2]的固体样品在10 K下的EPR谱;b1,K[2]和1-Pr在1kOe条件下的变温磁化率数据;c K[2]的SOMO和SOMO‒1轨道
为了进一步探究K[2],K[7]和K[8]的电子结构和成键性质,作者进行了基于密度泛函理论(DFT)的计算研究。DFT计算结果显示,[2]‒的SOMO和SOMO‒1轨道为近简并的铈4f轨道和底座芳烃π*轨道的δ相互作用,且铈的4f轨道占比要高于底座芳烃2p轨道的占比。这一结果表明对二价铈配合物电子结构的最恰当表示是通过δ反馈键稳定的4f2电子构型,与文献报道的大多数非传统二价稀土离子4fn5d1的电子构型迥异。K[2]特殊的4f2电子构型与配合物结构有关:在近似C3v的配位场下,配体底座芳烃的π给电子性大幅抬高了dσ轨道的能量,而dπ和dδ轨道则参与了与配体胺基氮原子的成键,因此铈的5d轨道能量过高无法填充电子;另一方面,还原后铈与底座芳烃的距离显著缩短,在小幅抬高fσ轨道能量的同时,通过铈与底座芳烃的δ成键作用大幅降低了fδ轨道的能量(图5a)。这一系列因素最终促使二价铈离子采用4f2的电子构型。此外,作者还对四价铈的末端氧配合物K[7]和末端亚胺配合物K[8]的铈‒氧和铈‒氮多重键性质进行了自然局域分子轨道(NLMO)的分析。分析表明,在铈‒氧三重键中,两个近简并的π键能量显著低于σ键,这在多重键中并不常见,之前仅见于铀酰等少数例子中,凸显了铈‒氧多重键的强度。与之相反,在铈‒氮多重键中,σ键能量远低于π键,而且计算得到的Wiberg键级1.23也显著低于铈‒氧键的1.58。
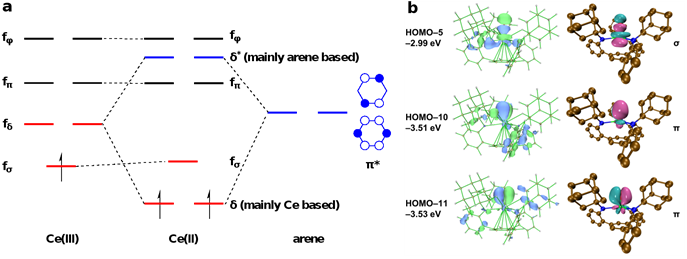
图5:a还原前后价层轨道能量和电子排布的变化;b四价铈末端氧配合物的铈氧π4σ2三重键
综上,该项工作合成了一系列三脚架型三胺基芳烃配体支撑的铈(II‒IV)配合物,并首次实现了稀土元素的单中心两电子氧化。实验表征和理论计算共同表明,二价铈配合物的电子结构为δ反馈作用稳定的4f2电子构型。此外,成键分析揭示了四价铈末端氧和末端亚胺配合物的铈‒氧和铈‒氮键多重键的特征。这些研究成果开启了稀土氧化还原化学的新篇章,也展示了“金属‒芳烃协同作用”这一策略在稳定多种氧化态和构建金属多重键方面的广阔前景。
该论文的第一作者为北京大学化学与分子工程学院博士研究生王怡,博士研究生梁杰锋、邓翀、孙荣、付鹏翔为该工作做出了贡献,王炳武研究员和高松教授为共同作者,黄闻亮研究员为通讯作者。该研究得到了国家自然科学基金、国家重点研发计划、北京大学、北京分子科学国家研究中心等的资助,并得到了北京大学分析测试中心、北京大学高性能计算平台等的支持。

